
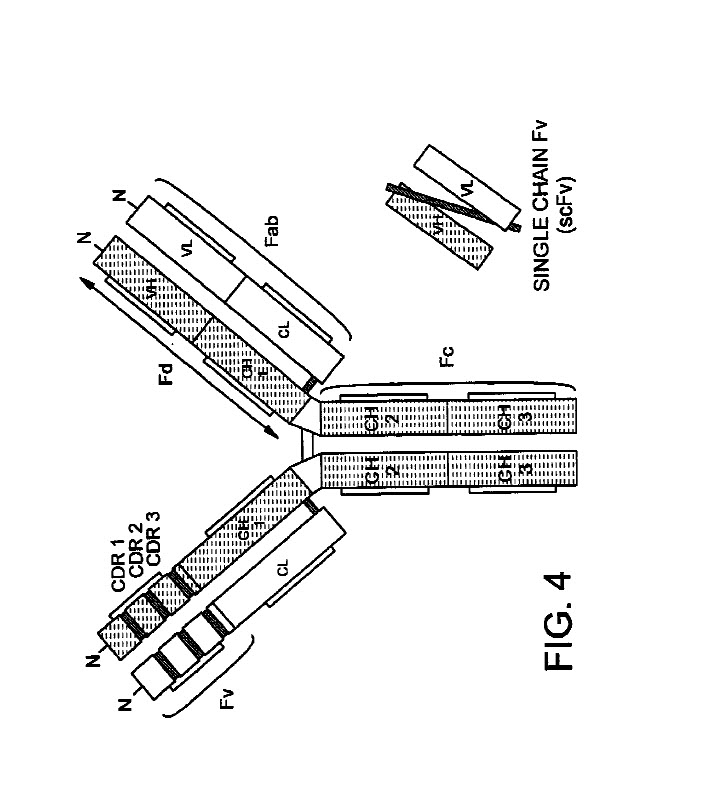
Figure from Nilsson (US6,740,734B1)
Introduction:
It is well documented that the structure of an immunogloublin forms a Y-shaped glycoprotein that is composed of two identical heavy and two identical light chains. Variable regions are assembled form two genes (V and J, for alpha and kappa light chains) or three genes (V, D and J for heavy chains), following the F(D)J recombination mechanisms. The joined regions are part of CDR3. Further variability in CDR3 lenght and sequence is introduced by the mechanisms that permit addition or deletion of nucleotides in those junctions and by somatic hypermutations in the recobmined genes. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018)
The antibody structural unit is known to comprise a tetramer. Each tetramer is composed of two identical pairs of polypeptide chains, each pair having one “light” (about 25kD) and one “heavy” chain (about 50-70kD). Thus naturally occurring antibodies (immunoglobulins) comprise 2 heavy chains linked together by disulfide bonds and two light chains, one light chain being linked to each of the heavy chains by disulfide bonds. Each heavy chain has at one end a variable domain (VH) followed by a number of constant domains. Each light chan has a variable domain (VL) at one end and a constant domain at its other end; the constant domain of the light chain is aligned with the first constant domain of the heavy chain, and the light chain variable domain is aligned with the variable domain of the heavy chain. See Goswami (Antibodies 2031, 2, 452-500)
Thus antibodies are composed of four polypeptide chains (two heavy chains of MW of about 50 kDa each, and two light chains of molecular weight of about 25 kDa each. The heavy chain contains four structural domains; 3 constant domains (CH1, CH2 & Ch3) and 1 variable domain (VH). The light chain is composed of two structural domains; one constant (CL) and one variable (VL). Based on the sequence of the constant region of the H chain, antibodies are divided into five classes: IgM, IgD, IgG, EgE and IgA. IgGs are further classified as IgG1, IgG2, IgG3 and IgG4, in the order of their relative abundance in plasma. Each of these also contains different heavy chains: gamma1, gamm2, gamma 3, and gamma 4, respectively. (Goswami, Antibodies, 2013, 2, 452-500, 2013).
A normal antibody heavy or light chain has an N-terminal (NH2) variable (V) region, and a C-terminal (COOH) constant (C) region. The heavy chain variable region is referred to as VH and the light chain variable region is referred to as VL. The amino-terminal portion of each chain includes a “variable” (“V”) region of about 100 to 110 amino acids primarily responsible for antigen recognition. The carboxy-terminal portion of each chain defines a constant region primarily responsible for effector function. The variable region is the part of the molecule that binds to the antibody’s cognate antigen, while the Fc region (the second and third domains of the C region) on the heavy chain determine the antibody’s effector function. Light chains have a constant region about 110 amino acids long and a variable region of the same size. The variable region of the heavy chains (at their N-terminus) is also about 110 amino acids long, but the heavy chain constant region is about 3-4 times longer depending on the class. Full-length immunoglobulin or antibody light chains are encoded by a variable region gene at the N-terminus and a kappa or lambda constant region gene at the COOH-terminus. Full-length immunoglobulin or antibody heavy chains are also encoded by a variable region gene and one of the constant genes (e.g., gamma).
For the heavy chain variable region, the hypervariable region ranges from amino acid positions 31-35 for CDR1, 50-65 for CDR2, 95-102 for CDR3. For the light chain varigion, the hypervariable region ranges from amino acid positions 24-34 of CDR1, 50-56 for CDR2 and 89-97 for CDR3. This numbering is in accordance with “Kabat labeling” used to number amino acid reisudes which are more variable (Kabat (1971) Ann. NY Acad, Sci. 190: 382)
The structure of the immunoglobulin molecule is determined by its primary, secondary, tertiary, and quaternary protein structure. The primary aa sequence accounts for the variable and constant regions of the H and L chains. The secondary structure is formed as the chain folds back and forth upon itself forming an antiparallel ? sheet which is stabilized by an intrachain disulfide bond and by hydrogen bonds that connect the peptide bonds in neighboring chains. The chains fold into a tertiary structure of compact globular domains. These domains of adjacent H and L chains interact in the quaternary structure to form domains that enable the molecule to specifically bind antigen.
It is estimated that 30 residues underlying the CDRs, 16 in VH and 14 in VL, are responsible for stabilizing the HV loop structure as well as modifying their positioning. Since these residues fine-tune the antibody affinity, this region is called “Vernier Zone”. (Fransson “Humanization of antibodies” Frontiers in Bioscience 13, 1619-1633, 2008)e basic strcuture of IgG is composed of two light chains and two heavy chains to form a complex quaternary Y shaped structure with ghree independent protein moieties connected thorugh a flexible hinge region. These moieties are symmetrical with two identical fragment antigne-specific binding (Fab) regions and one fragment crystallizalbe (Fc) region. (Wang, Design and Production of Bispecific Antibodies” Antibodies, 2019).
Th
Fab: “Fab” or “fab region” denotes the VL, VH, CL, and Ch1 of the immunoglobulin domains or regions. (Zhang (WO 2017/034770).
Heavy Chain (HC):
Light Chain (LC):
Two types of light chain, termed lambda and kappa are found in antibodies. A given immunoglobulin either ahs kappa or lambda chains, eever one of each. No functional difference has been found between antibodies haivng lambda or kappa light chains. (Himmler US 2010/0184615)
Human immunoglobulin light chains (LC) consist of two domains, each of 100-110 amiino acids (12 kDa, approximately), that fold independently of each other. The variable region domain (VL), generated via rearrangement of a V to a J gene segment, and the constant region domain (CL) of minimal variation. LC can occur in two isotypes, namely kappa and lambda. The two LC domains have similar compact tertiary structure with a hydrophobic core composed of two twisted beta-sheets, which are stabilized by a single intrachain disulfide bond. On the other hand, a cysteine (for a total of 5 cysteines in a single LC molecule) located at the carboxyterminal end, forms an interchain disulfide bond with the immunoglobulin heavy chain. LC are usually synthesized in large excess over H chains, and this results in production of “free” LC that an be detected in serum and urine. Kappa free LC are normally monomeric and can be found with various degrees of glycosylation, while lambda free-light cahins tend to be dimeric and unglycosylated. (Rognoni, A strategy for synthesis of pathogenic human immunoglobulin free Light chains in E. coli.” PLOS one, 8(9), 2013).
VL Domain
The VL domain provides contacts critical to antigen binding. It is unlikely that the affinity of isolated VH fragments will generally match that of intact antibodies. The absence of the VL domain leaves a large hydrophobic patch on one face of the Vh fragment, which will amost certainly lead to increased nonspecificity relative to whole antibodies (Huse, “Generation of a large combinatorila library of the immunoglobulin repertoire in phage lambda” Science, 1989, 246, pp. 1275-1281 at p. 1276).
In vivo, human VL constructs are the result of genetic recombination between germline gene segments VL and JL. The first two complementarity-determining regions (CDR1 and CDR2) and a part of the CDR3 up to residue 95 are encoded by VL segment genes; the fo the CDR3 and the entire framework region (FR) 4 areencoded by JL gene segments. Human VL are classified as eith kappa or lambda subtypes, with seven Vkappa gene segment subgrounds and 11 Vlambda gene segments. (Kim “Antibody light chain variable domains and their biophysically improved versions for human immunotherapy” mAbs , 219-235, 2014).
In general, VH domains exhibit higher solubility and stability that V kappa domains, possibly due to a higher packing density in their upper core and a more hydrophilic C-terminus, and among the Vkappa subgroups, the Vkappa3 subgroup members exhibit the best properites in terms of solubility and thermodynamic stability. A significant. proporition of human VL, prepdomenintantly of the Vkappa class, bind to the B cell super-antiogen protein L. Kim “Antibody light chain variable domains and their biophysically improved versions for human immunotherapy” mAbs , 219-235, 2014).
Fc Domain (see outline)
Disulfide Bonds
One of the basic strucural features of human IgG1 is the arrangement of the disulfide bond structure; 4 inter chain disulfide bonds in the hinge region and 12 intra chain disulfide bonds associated with twelve individual domains. Disulfide bond structure is critical for the structure, stability, and biological function of IgG molecules. It is known that inter chain disulfide bonds are more susceptible to reduction than intra chain disulfide bonds. In addition, the disulfide bonds between the light chain and heavy chain are more susceptible than disulfide bonds between the two heavy chains. The upper disulfide bond of the two inter heavy chain disulifde bonds are also mroe susceptible than the lower one. In addition, disulfide bonds in teh CH2 domain are the most susceptible to reduction. (Liu “Ranking the Susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis” Analytical Chemistry, 92(12), 2010).
Disulfide bonds in the hinge region connect the two heavy chains. The light chains are coupled to the heavy chains by additional disulfide bonds. Asn-linked carohydrate moieties are attached at different positions in constant domains dependeing on the class of immunoglobulin. For IgG1 two disulfide bonds in the hinge region between Cys235 and Cys238 pairs, unite the two heavy chains. The light chains are coupled to the heavy chains by two additional disulfide bonds beetween Cys229s in the CH1 domain and Cys214s in the CL domain. Carbohydrate mooieties are acched to Asn306 of each CH2, generating a prounounced bulge in the step of the Y. (Himmler US 2010/0184615)
How amino acids are numbered
Kabat Numbering Scheme:
In 1970, Kabat and Wu aligned 77 Bence-Jones protein and immunoglobulin light chain sequences in order to study the statistical variability in amino acid composition at the sequential positions of the variable regions. The analysis revealed three hypervariable regions in the variable region of the L chains. The presence of highly conserved residues was also demonstrated, such as the two csteines that form a disulphide bridge at the inner core of the immunoglobulin domain and a tryptophan residue located immediately after CDRL1. In 1979, Kabat were the first to proposed a standardized numbering scheme for the V region. In their compilation of “Sequences of Proteins of Immunological Interest”, the amino acid sequences of the V region of the L (alpha, kappa) and H chain of antibodies as well as the variable regions of T cell receptors (alpha, beta, gamma, epsilon) were aligned and numbered. Although the Kabt nubmeirng scheme is often consdiered as the standard that is widely adopted for numbeirng antibody residues, it has some important limitations. First, this scheme was built on the alignments of a limtied number of sequences form antibodies with the most common sequence lenghts. Consequently, sequences with unconventional insertions or deltetions in the CDs or inthe framework regions were not included. Thus, the Kabat scheme ignores antibody chains of unconventional lenghts with unique insertions or deletions. However, a useful numbering tool named ABnum that numbers the amino acid sequences of V domains according to a much larger and regularly updated database (Abysis) takes into account insertions of varialbe lenghts, particularly in CDR2 by adding an insertion point at position L54. The second main limitation of the Kabat sceeme is that it does not match very well with the 3D structure of antibodies. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018)
The Kabat numbering system is generally used when referring to a residue in the variable domain (approximately residues 1-107 of the L chian and 1-113 of the H chain). The EU numbering system or “EU indeix” is generally used when referring to a residue in an immunoglobulin heavy chain contant region. The “EU index as in Kabat” refers to the residue numbering of the human IgG1 EU antibody. (Yeung WO 2010/075548).
Chothia Numbering Scheme:
In 1987, Chotia and Lesk introduced a structure based nubmeirng sheme for antiboy V regions. They aligned cyrstal structures of antibody V regions, defined the loop structures that form the CDRs and corrected the position numbers of the insertion points inside CDRL1 and CDRH1 so that they better fit their topological positions. Furthermore, they classified the CDR loops of H and L chains in a small number of conserved structures, called “canonical” classes. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018)
Chothia and Lesk define the “hypervariable loops” based on the relationship between amino acid sequences and three-dimensional structures around the antigen binding region. They found that 5 out of 6 hypervariablere region (i.e., light chain CDR loop 1-3(L1,L2,L3) and heavy chain CDR loop 1-2(H1 and H2) typically adopt a limited number of discrete backbone conformations, called “canonical structures”. They further identified several residues that are responsible for the main chain conformations of the hypervariable loops. To build the reliable antibody variable fragment (Fv) structures, people have extensively studied the conformational library of canonical structures. Different from the canonical structure approach, ab initio methods dpend on physicochemical principles rather than the template. Due to the simplificaiton of the energy functions and the limited computational resources, the ab initio methods have limitation in accuracy. (Zhao, “In silico methods in antibody design” Antibodies, 2018)
Martin Numbering Scheme:
In 2008, Martin focussed on the structural alignment of different framework regions of unconventional lenghts. They highlighted residues that are absent in most sequences and structures and thus defined these as deletion positions. By analyzing sequences and structures, they also proposed a correction of the insertion point within the framework region 3 of the H chain domain from position H82 to H72. In addition and by analogy with CDRH2, they amended the position of the insertion point for the CDRL2 that locates now at position L52. Finally, they used the nubmering software, Abnum, and recommended a new numbering shceme that consists of the Chothia numbering system corrected by teh ABnum software. This software uses the much larger Abysis database, which integrates sequences form Kabat, IMGT and the PDB databases. For this reason, the ABnum program defines a noval insertion/deletion position at position H6 int eh Chothia and Kabat numbeirng schemes. The Martin numbering scheme corrects this point of insertion and shifts it toward positions H8. The Martin numbering shceme should be consdiered as the most recent version of the Chothia numbering. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018).
Gelfand Numering Scheme:
This numbeirng system results in a realtively complex nomenclature. The V chain sequences are divided into 21 fragments termed “words”, each of these “words” matches with a secondary structure element (a strand or a loop). The strands are defined by a letter in alpahbetical order (e.g., A, B, C) and the loops by two letters that corresponds to the neighboring strands (e.g., AB, BC…) However, there are two exceptions in this terminology: the three N-tmerinal residues of the V chains and the loop connecting the B and C strands, which as a two span bridge conformation with one residue deeply inserted into the structure. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018)
IMGT Numbering Scheme:
In 1997, Lefrance introduced a new and standardized numbeirng system for all the protein sequences of the immunoglobulin supermafily, including V domains form antibody L and H chains as well as T cell receptor chains fro different speies. The shceme was based on amino acid sequence alignment of the germ line V genes. Consequently, the amino acid sequence and numbeirng stops wehre CDR3 should start. Later on, the authors extended their scheme to the entire V domains and developed various tools to analyze the full lenght sequence. IMGT possesses its own defintions of the framework regions (named FR-IMGT) and CDR (named DR-IMGT). The IMGT numbering method coutns resiudes continuously form 1-128 based on the germ line V sequence alginment. Thus, it avoid the sue of insertion codes, except between position 111 and 112 for CDR3-IMGT with mroe than 13 amino acids. Conversely, no number is attributed when a resiude is missing in a particular sequence. For example, in a 6 amino acid long CDR1-IMGT, residue 27 is followed by residue 34 (and residues 28-33 are absent). The scheme is based on alignments of sequences form an immunoglobulin superfamily. This has led to the development of highly useful tools. For example, amino acid alignment and numbering can be performed by the IMGT/DomainGapAlign tool. (Dondeliner “Understanding the significance and implications of antibody numbering and anitgen-binding surface/residue definition” Frontiers in Immunology Volumne 9, 2018)